I ricercatori hanno utilizzato lo strumento CRISPR gene-editing per manipolare il modo in cui bobine di DNA si posizionano all’interno della cellula – un altro passo della ricerca per capire come impatta e funziona la struttura del genoma
Quando gli scienziati finirono di sequenziare il genoma umano nel 2003, molti ricercatori si concentrarono sulla decodifica delle lunghe sequenze di As, Ts, GS e Cs per comprendere i misteri della genetica umana. Il genoma, tuttavia, non appare in natura come una semplice lunga serie di lettere. Srotolato, si estende per circa due metri, ma si piega e avvolge in rotoli per riuscire ad adattarsi all’interno di un nucleo che ha un diametro inferiore a 10 micron.
Negli ultimi anni, i ricercatori hanno cominciato a capire quanto questa architettura genetica 3-D sia importante. Proprio come le uniche informazioni accessibili in un libro sono lì sulla pagina aperta di fronte a voi, le istruzioni del genoma possono essere lette solo quando tali istruzioni non sono nascoste all’interno di profonde pieghe. Ma i ricercatori non capiscono come la cellula pieghi il DNA in modo tale da riuscire poi a leggerne le parti importanti.
All’inizio di quest’anno, due gruppi indipendenti di ricercatori hanno fatto un grande passo avanti verso la decodifica dei misteri del ripiegamento del DNA. Entrambi i gruppi hanno utilizzato il nuovo e potente strumento di gene-editing, noto come CRISPR, per srotolare un segmento del DNA. Il loro lavoro potrebbe aiutare gli scienziati a identificare alcune delle regole di base che stanno dietro il come e perché il genoma si costituisca in una struttura 3-D. Esso rivela anche che la presenza di piccole sequenze di DNA possono comportare enormi variazioni su come sia organizzato il genoma.
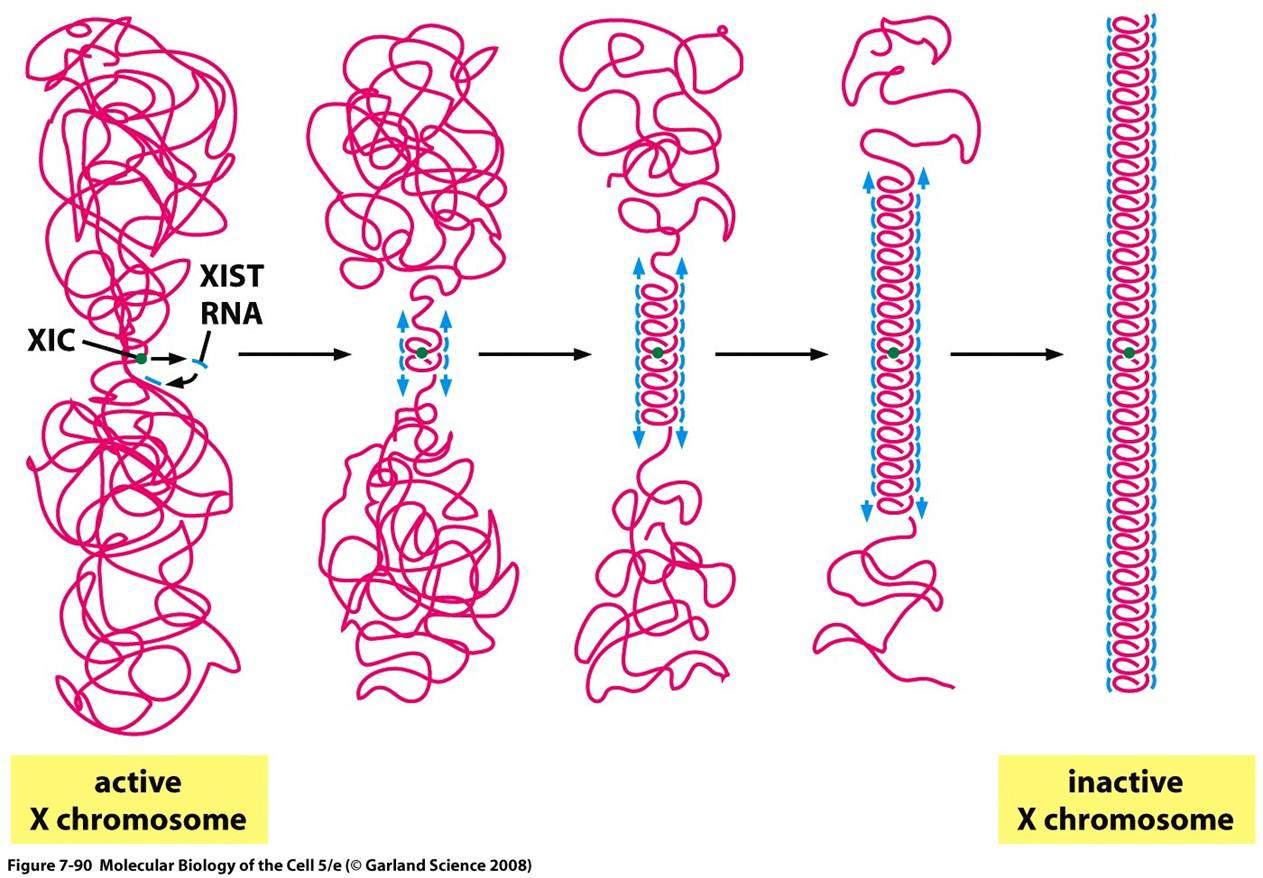
La nuova ricerca ha debuttato con un primo tentativo: comprendere un mistero del cromosoma X. La maggior parte dei mammiferi maschi ha un cromosoma X e uno Y, mentre la maggior parte delle femmine ha due cromosomi X. Questo costituisce un potenziale problema nel meccanismo cellulare di una femmina. Se entrambi gli X rimangono attivi, il doppio del numero di geni del cromosoma X sarà attivo. Questo porterà a una serie di difficoltà di sviluppo, che spesso si conclude con la morte dell’embrione subito dopo la fecondazione.
Per evitare questo scenario, una copia del cromosoma X si spegne con un gene chiamato Xist. Il cromosoma X inattivo, noto come il corpo Barr, è piccolo. Si arrotola e nasconde quasi tutti i suoi geni dal meccanismo cellulare che trasforma il DNA in RNA e proteine.
“L’inattivazione di un X è un processo critico dello sviluppo femminile”, ha dichiarato Brian Chadwick, biologo presso la Florida State University. “L’unica differenza tra i due X è come vengono confezionati.”
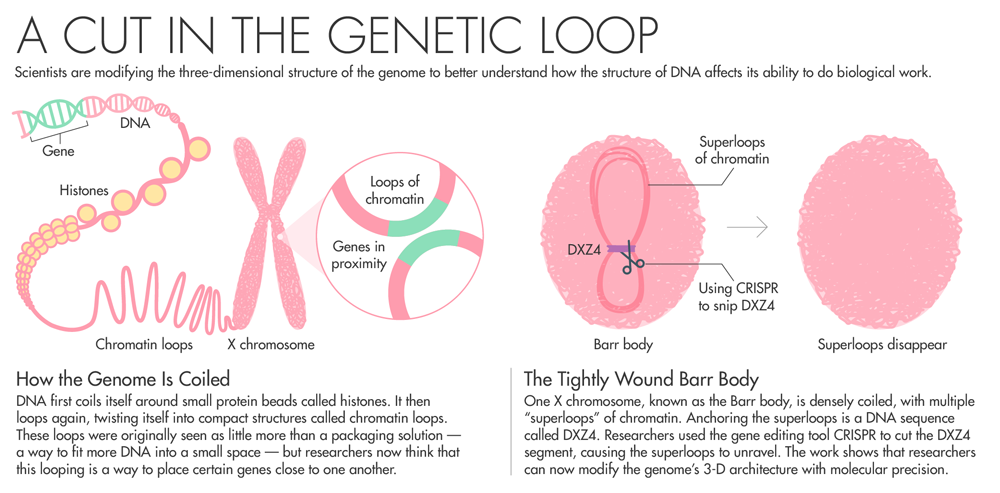
Chadwick ha trascorso tutta la sua carriera cercando di capire come il corpo inattivi una copia del cromosoma X. Il lavoro di Chadwick sui corpi Barr si è concentrato su una sequenza di DNA chiamato DXZ4 come potenziale chiave del ripiegamento che disattiva la maggior parte dei geni di una copia del cromosoma X. La sua squadra ha controllato come un cromosoma X venga disattivato nelle sequenze di tutti i mammiferi.
Ma come ha fatto Chadwick a verificare se DXZ4 era veramente il maestro di origami del cromosoma X? Un importante indizio è apparso nel 2014, nel lavoro che aveva cominciato più di due decenni prima.
I primi collegamenti
Come studente di dottorato nel laboratorio di Mark Seyfred, alla Vanderbilt University, agli inizi degli anni 1990, Katherine Cullen voleva capire come una proteina chiamata prolattina, permettesse alle femmine dei mammiferi di indurre la produzione di latte. Cullen e Seyfred credevano che l’esposizione ad estrogeni innescasse una serie di eventi a cascata che terminava con la creazione di un ciclo gigante di DNA. Questo ciclo si collega ed è il promotore del gene prolattina, che agisce come un interruttore per attivare il gene, con una sequenza nucleotidica prolattina, che funziona come un dito metaforico che solleva l’interruttore. Il compito del dottorato di Cullen era quello di verificare questa condizione.
Nel genoma, “nulla ha senso se non in 3-D.”
E-Coli.
Per cercare questo ciclo usò una tecnica, che è stata recentemente sviluppata da Seyfred, chiamata proximity ligation. Il primo passo è quello di trattare il genoma con formaldeide, che crea legami incrociati tra i segmenti di DNA che sono vicini l’uno all’altro. Il processo rivela quali parti del genoma tocchino anche se tali sequenze sono lontani l’uno dall’altro sul genoma lineare. Cullen aveva scoperto che l’esposizione ad estrogeni crea un ciclo che collega la sequenza nucleotidica prolattina e il promotore. La sua scoperta le valse una pubblicazione nel 1993 su Science.
Il lavoro di Cullen offrì alcune delle prime prove concrete che la più grande struttura tridimensionale del genoma è legata alla sua funzione. Ma il suo articolo non ricevette molta attenzione.
Le cose un decennio più tardi cambiarono, quando ricevette una telefonata a sorpresa da Erez Lieberman Aiden, della Harvard University, che era alla ricerca di ulteriori informazioni sul test di legatura che aveva usato. Aiden nutriva grandi speranze su questo test, che non voleva usare solo su un gene, ma su tutto il genoma, per identificare non una sola sequenza, ma potenzialmente migliaia di loro.
Nel corso dei prossimi anni, Aiden, che al momento è un biologo computazionale presso la Rice University e il Baylor College of Medicine, basandosi sul lavoro di Job Dekker, un biologo presso Medical School University, del Massachusetts, sta creando il sistema Hi-C, che mappa la probabilità che due pezzi di genoma si tocchino.
“Ci imbattiamo nel nostro vicino di casa più spesso che con qualcuno che vive in un altro paese. L’idea è proprio questa”, ha detto Aiden. “Se sappiamo con chi si va a imbattere, in una normale giornata, il genoma, allora possiamo capire quanto gli sono vicino queste aree e di conseguenza ciò che al genoma si presenta come in tre dimensioni.”
Aiden e colleghi hanno pubblicato i risultati dei loro primi esperimenti Hi-C nel 2009. Questi risultati hanno rivelato l’architettura del genoma con una risoluzione di 1 milione di paia di basi – uno schema che ha cominciato a rivelare la complessità incredibile del genoma. “I risultati sono stati come una mappa del mondo che ti ha mostrato solo i perimetri sfocati dei continenti”, ha detto Suhas Rao, uno studente laureato che lavora nel laboratorio di Aiden. “Non si poteva davvero navigare con loro, ma è stato un punto di partenza.”
Il laboratorio Aiden ha trascorso gli anni successivi a raffinare il sistema Hi-C e, nel 2014, ha pubblicato un documento con ogni singolo tracciato ad anello e la bobina del DNA a una risoluzione di 1000-base-pair. Se la mappa del 2009 ha rivelato dei perimetri sfocati del Nord America, il nuovo Hi-C ha mostrato perfino una grata sull’asfalto di una strada di Manhattan. Dettaglio che ha dato agli scienziati i primi indizi sulle regole con le quali il genoma si ripiega.
Di tutti i miliardi di contatti tra DNA che di Aiden Hi-C ha generato, una zona del genoma si è distinto. Nelle cellule femminili XX, Rao, Aiden e colleghi hanno scoperto che, mentre una X segue un modello ad anelli condiviso con il resto dei cromosomi, gli X inattivi sembrano molto diversi. Invece di avere più sequenze di circa 200.000 paia di basi, gli inattivi X avevano due enormi “superdomini” caratterizzati da molteplici “supersequenze” fino a 77 milioni di coppie di basi. Qual’è stato l’ancoraggio delle supersequenze? Una sequenza di DNA chiamata DXZ4 – che lo stesso Chadwick aveva precedentemente identificato come chiave per la piegatura del cromosoma X. Chadwick legge il giornale e decide di aiutare Aiden ed è così che i due decidono di collaborare.
Un taglio genetico
Comprendere il rapporto tra la struttura di una molecola e la sua funzione è una delle classiche domande della biochimica. Gli scienziati che studiano le proteine hanno imparato che questo campo di indagine, a partire dagli anni 1960, sta cambiando il concetto di aminoacido, il mattone di una proteina, e misurando come si modifica la capacità di una proteina nel fare il suo lavoro. Chadwick e Aiden volevano fare qualcosa di simile a comprendere la relazione tra le sequenze di DNA e l’architettura del genoma. Come molti laboratori di genetica, si rivolsero allo strumento CRISPR, che agisce come un insieme di forbici biomolecolari, per modificare il genoma.
Per dimostrare che DXZ4 influenza realmente il ripiegamento del genoma, il team ha preso delle cellule umane e utilizzato CRISPR per tagliare la sezione DXZ4 – un tratto ripetitivo del DNA che si ripete per centinaia di migliaia di nucleotidi. Hanno quindi utilizzato Hi-C per misurare quanto il taglio avesse influenzato la sequenza del cromosoma e quando hanno tolto DXZ4, “quei cicli giganteschi erano scomparsi. Il cromosoma comincia ad assomigliare ad un autosoma normale “, riferisce Aiden. “E ha dimostrato che avremmo potuto avere un controllo preciso su come si ripiega il genoma.”
Per conto suo, il laboratorio di Dekker aveva parimenti dimostrato il ruolo chiave del DXZ4 nel ripiegamento del cromosoma X inattivo dei topi, rilevando inoltre che il gene Xist – l’interruttore molecolare che arrotola il cromosoma X inattivo – contribuisce a creare il confine tra i due grandi superdomini di quel cromosoma. Sia la mappa di Dekker (pubblicato su Nature) sia Aiden e Chadwick (pubblicati negli Atti della National Academy of Sciences) hanno contribuito a districare il nodo gordiano del genoma pieghevole.
“E ‘abbastanza spettacolare che la struttura di un intero cromosoma possa contare su una piccola sequenza di DNA da qualche parte nel mezzo”, ha detto Wouter de Laat, un genetista biomedico presso l’Università di Utrecht, nei Paesi Bassi.
La nostra conoscenza sul rapporto intimo che esiste tra il modo in cui si piega il genoma e su come funziona, ha riferito de Laat, si sta espandendo. Gli scienziati hanno a lungo sospettato che un’anormale ripiegamento del genoma può causare delle malattie, e molti nuovi studi hanno identificato dei legami tra l’architettura del genoma e il suo sviluppo biologico. Uno studio del 2016 di Stefan Mundlos, presso l’Istituto Max Planck, di genetica molecolare a Berlino, ed i suoi colleghi hanno dimostrato che un riarrangiamento del DNA in una regione non codificante del genoma ha causato malformazioni degli arti durante lo sviluppo, cambiando la ripiegatura della cromatina. Altri ricercatori stanno utilizzando CRISPR per verificare se le modifiche dell’architettura del genoma influenzi la capacità di parassiti come Trypanosoma, di causare la malattia del sonno africana, per eludere il sistema immunitario.
Come dice Dekker, “nulla ha senso se non è visto in una prospettiva tridimensionale.”
A cura di Adriana Paolini
Linkografia
https://www.youtube.com/watch?v=2pp17E4E-O8
http://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.1000276